Introduction
Established methods for computational phylogenetics were employed. We used BLAST+, Hmmer3, MUSCLE and MEGA for generating alignments (and tree construction), see full list and links (at bottom of page).
Raw data was provided to the consortium members around March 2013. By June 2013, a complete dataset had been downloaded and computational methods established.
MAAB Pipeline
The motif and amino acid bias (MAAB) bioinformatics pipeline classifies HRGPs into 23 descriptive sub-classes, as illustrated in below: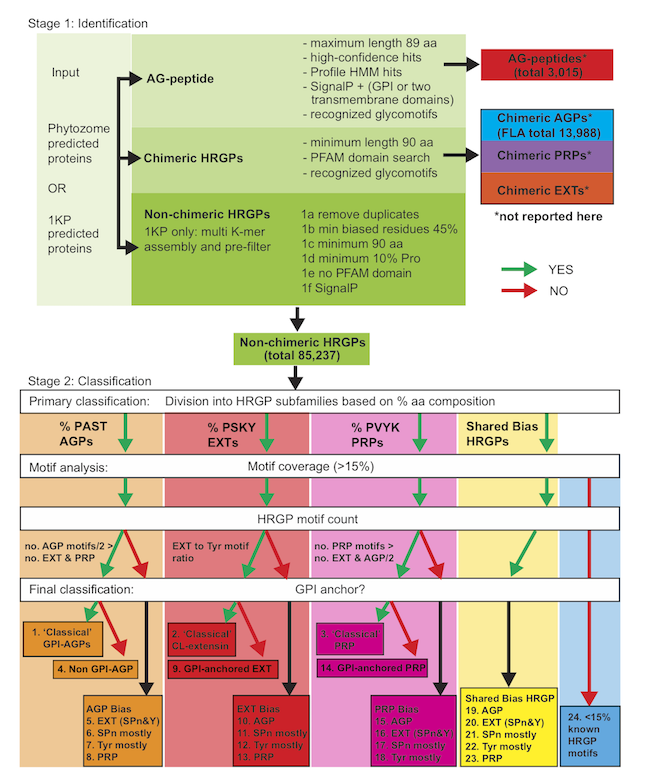
Figure 3, Johnson et al. (2017a).
Tools employed
The following software were used in developing the MAAB pipeline and in subsequent analysis.| Software package | Comments |
|---|---|
| KNIME | Tools developed for the MAAB pipeline, together with existing tools, were joined into workflows using the KNIME analytics platform. |
| TRAL | We applied tandem repeat analysis to the MAAB output. Summarised results and search are available. |
| MSA Viewer | Used for displaying alignments on the TRAL search tool page. |
| NCBI BLAST+ | Used for identification of similar sequences for numerous gene families |
| Hmmer v3.1b1 | Used for identification of TRAL-identified tandem repeats and AtAGP6/11 putative orthologues |
| SignalP v4.0 | Used for prediction of the presence of N-terminal ER signal sequence. The "noTM" model was specified as this gave a more accurate results based on existing knowledge of plant HRGPs |
| BigPI plant predictor | Used for detection of the presence of C-terminal GPI-anchor signal sequences. We obtained a local copy of the server (thanks to Eisenhaber et al.), and employed this software in large scale to identify GPI anchors in key gene families. |
| Oases | Used to reassemble the transcripts from RNA-seq read data from 1KP (four k-mers: 39, 49, 59 and 69) to better span tandem repeats that are common to several types of HRGPs (EXTs and PRPs). |
| EMBOSS getorf | Used to provide open reading frame predictions from reassembled transcriptomes (using oases, k= 39, 49, 59 and 69). A BioPerl script is available that was used to identify putative ORFs translated into amino acids using getorf. |
| Usearch | Used to cluster similar sequences. |
| MEGA6 | The best model for each alignment (MUSCLE) was identified and used to generate a maximum likelihood tree (with 100 bootstrap replications), using all sites in the alignment |
Code
The core stage 2 classification code for MAAB, as described in Methods and applied to Phytozome and 1KP data, is available below as Java source code. This can be used as a reference to implement and adapt the MAAB classifications in a programming language of choice, and to integrate it into existing analysis workflows where required filtering for stage 1 is already implemented.
Additionally, to encourage the use and adaptation of MAAB, a generalised implementation of the pipeline for HRGPs, MAAB Template, is available for download. It can be used in an iterative approach, to allow refinement to suit specific datasets/sequences of interest , and to inspect the individual steps of MAAB.
Suggestions as to how to modify MAAB for HRGPs are provided in Johnson et al. ‘A motif and amino acid bias bioinformatics pipeline to identify hydroxyproline-rich glycoproteins’ (in press). MAAB may also improve the recovery and classification of other classes of intrinsically disordered proteins (IDPs) such as elastins, salivary PRPs, insect silks, AGL proteins from arbuscular mycorrhizal fungi, and LATE EMBRYOGENESIS ABUNDANT (LEA) proteins in plants.
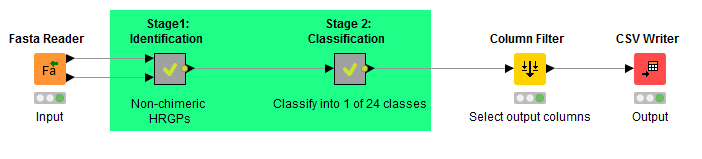
MAAB pipeline template implemented in KNIME.
| Source | Comments |
|---|---|
| MAAB Template | The KNIME workflow has been refined from the highly customised 1KP and Phytozome analysis, replacing some unique steps to analyses with simpler alternatives. It also omits components that have software license restrictions (e.g. BIG-PI Plant Predictor, Eisenhaber et al 2003). MAAB Template takes protein sequence data in FASTA format as input, and classifies each sequence into one of 24 MAAB classes. This pipeline is implemented as a KNIME workflow (.knwf file) and requires the KNIME Analytics Platform (https://www.knime.org/). |
| MAAB Stage 2 | The key code in MAAB Template for classifying HRGPs. We developed a decision tree pipeline (motif and amino-acid bias (MAAB)), implemented in Java, to filter sequences for indicative HRGP compositional biases (AGP, EXT, PRP) and then into one of twenty-four MAAB classes (23 HRGP classes and a final MAAB class, with <15% known HRGP motifs). |
| Large dataset pre-filter for Stage 1 | A BioPerl script is provided to scan the translated ORFs found in each transcriptome to identify sequences which are at least 90 amino acids in length, compositionally biased (%PAST/%PSKY/%PVYK >= 45), at least 10% proline (to create Input data for Stage 1 of MAAB). |
| RoI | A simple algorithm to identify proline-rich regions-of-interest (RoI) in protein sequences, implemented in Java. The percentage of the sequence covered by such biased regions is designated RoI% as a fraction of the total sequence length, i.e. including all signal sequences, if present. RoI% is one of the parameters reported in MAAB output |
| XP Motif | Script to identify glycomotifs in proteins of the form XPn for X = all 20 amino acids, and n = 1 through 5. |
National Collaborative Research Infrastructure Strategy (NCRIS)
This research was enabled by the following NCRIS supported projects:
- Nectar Research Cloud, a collaborative Australian research platform (https://nectar.org.au/)
- Research Data Storage Infrastructure (RDSI) Project (https://www.rds.edu.au)
- Bioplatforms Australia Pty Ltd (http://www.bioplatforms.com/)
Acknowledgements
The huge collective effort of the authors of the above software packages is wonderful and it has been a delight to use these projects. Demanding, but delightful none-the-less. A huge thank you to all of you who make larger-scale work possible in a timely fashion.